
Mitochondria dysfunction and Fanconi syndrome
Inborn disorders of energy provision by mitochondrial respiratory chain are the primary cause of numerous serious diseases, ranging from most severe encephalo-cardio-myopathies manifesting early after birth to various tissues-specific and milder disorders affecting mainly adults.
Underlining genetic defects can affect mitochondrial genome (mtDNA) or a number of nuclear genes. In case of isolated deficiency of one complex of respiratory chain, either some of the structural subunits or a biogenetic factor essential for enzyme biogenesis and assembly can be affected.
Joint studies of researchers from Institute of Physiology CAS and from Institute of Inherited Metabolic Disorders of the 1st Faculty of medicine Charles University, which collaborate within the consortium MITOCENTER, led to the characterization of the genetic cause and molecular mechanism of Acadian variant of Fanconi syndrome, characterized by generalized proximal tubular dysfunction from birth, slowly progressive chronic kidney disease and pulmonary interstitial fibrosis. Based on exome and genome sequencing in affected families, we identified pathogenic mutation in noncoding region of NDUFAF6 gene encoding assembly factor of respiratory chain complex I, NAHD dehydrogenase. Analysis of pathogenic transcripts indicated that mutation leads to a loss of mitochondrial isoform of NDUFAF6 factor and subsequently to alteration of biogenesis and function of complex I. Pathogenicity of mutation was finally confirmed by complementation – rescue of enzyme defect in patient fibroblasts by transfection of wt NDUFASF6 gene. Elucidation of molecular pathogenesis of Acadian variant of Fanconi syndrome will improve diagnostics and prevention of the disease in affected families and significantly broadens our knowledge of mitochondrial diseases associated with complex I disorders.
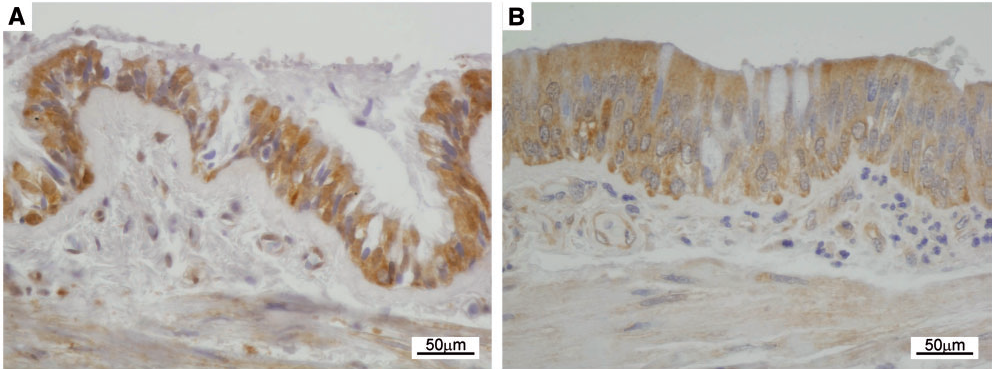
Immunohistochemical detection of NDUFAF6 expression in pulmonary epithelium of a control (A) and Fanconi syndrome patient (B) shows reactive epithelial changes and lower NDUFAF6 signal in the patient.
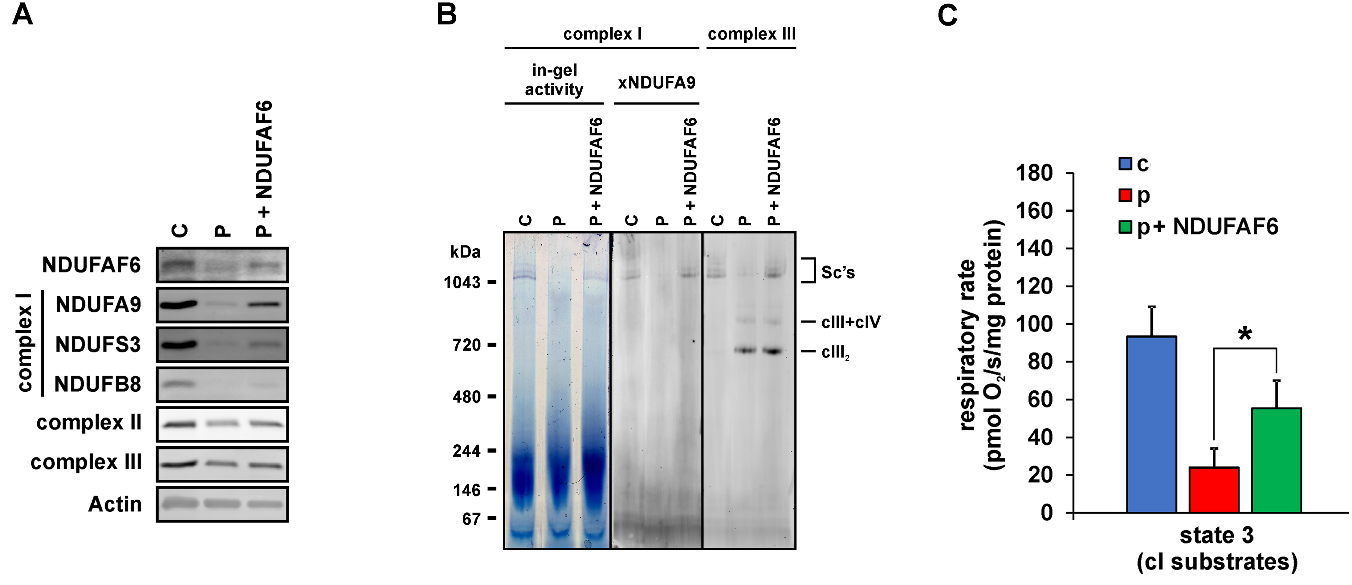
Complex I defect in fibroblasts of Fanconi syndrome patient is complemented by wt NDUFAF6.
A - Immunodetection of SDS-PAGE resolved NDUFAF6 protein, complex I subunits (NDUFA9, NDUFS3, NDUFB8), complexes II and III and actin in the fibroblasts from control (C), patient (P) and patient transfected with wt NDUFAF6 (P+NDUFAF6). B – Complex I activity and immunodetection of native complex I and complex III in digitonin-solubilised fibroblasts resolved by native electrophoresis (CNE or BNE). CIII2 denotes the dimer of complex III; CIII+CIV denotes the supercomplex of complex III and complex IV and SC’s denotes supercomplexes containing complexes I, III and IV. C – ADP-stimulated oxidation of complex I-dependent substrates (pyruvate+malate+glutamate) is decreased in patient (P) compared to control (C) and significantly restored upon transfection with NDUFAF6 (P+NDUFAF6). *P<0.05.
Hartmannová, Hana – Piherová, Lenka – Tauchmannová, Kateřina – Kidd, Kendrah – Acott, Phillip – Crocker, John – Oussedik, Youcef – Mallet, Marcel – Hodaňová, Kateřina – Stránecký, Vktor – Přistoupilová, Anna, Barešová, Veronika, Jedličková, Ivana, Zivná, Martina, Sovová, Jana, Hůlková, Helena – Robins, Vicki – Vrbacký, Marek – Pecina, Petr – Kaplanová, Vilma – Houštěk, Josef – Mráček, Tomáš – Thibeault, Yves – Bleyer, Anthony – Kmoch, Stanislav. Acadian variant of Fanconi syndrome is caused by mitochondrial respiratory chain complex I deficiency due to a non-coding mutation in complex I assembly factor NDUFAF6. Human Molecular Genetics. 2016, roč. 25(18), Sep 15, p. 4062-4079. IF = 5.985