
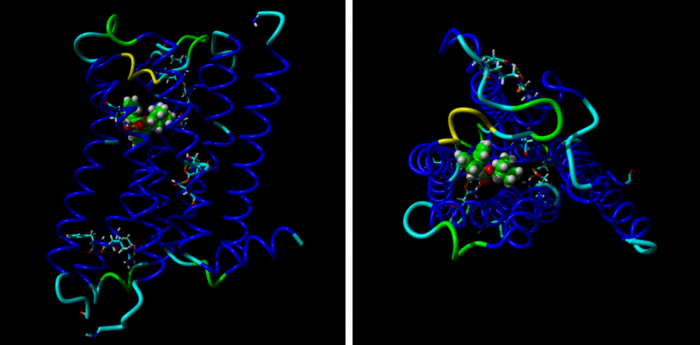
 Model M2 receptoru při pohledu z boku (vlevo) a z mimobuněčného prostoru (vpravo) ukazuje 7 modrých α helixů protínajících buněčnou membránu a navázaného antagonistu chinuklidinylbezilát (zelený).
Model M2 receptoru při pohledu z boku (vlevo) a z mimobuněčného prostoru (vpravo) ukazuje 7 modrých α helixů protínajících buněčnou membránu a navázaného antagonistu chinuklidinylbezilát (zelený).
Naše nová publikace: Homologní modelování muskarinového receptoru M2 pro acetylcholin
Model M2 receptoru při pohledu z boku (vlevo) a z mimobuněčného prostoru (vpravo) ukazuje 7 modrých α helixů protínajících buněčnou membránu a navázaného antagonistu chinuklidinylbezilát (zelený).
Jakubík J, Randáková A, and Doležal V (2013) J Comput Aided Mol Des 27: 525-538.
Abstract
Twelve homology models of the human M2 muscarinic receptor using different sets of templates have been designed using the Prime program or the modeller program and compared to crystallographic structure (PDB:3UON). The best models were obtained using single template of the closest published structure, the M3 muscarinic receptor (PDB:4DAJ). Adding more (structurally distant) templates led to worse models. Data document a key role of the template in homology modeling. The models differ substantially. The quality checks built into the programs do not correlate with the RMSDs to the crystallographic structure and cannot be used to select the best model. Re-docking of the antagonists present in crystallographic structure and relative binding energy estimation by calculating MM/GBSA in Prime and the binding energy function in YASARA suggested it could be possible to evaluate the quality of the orthosteric binding site based on the prediction of relative binding energies. Although estimation of relative binding energies distinguishes between relatively good and bad models it does not indicate the best one. On the other hand, visual inspection of the models for known features and knowledge-based analysis of the intramolecular interactions allows an experimenter to select overall best models manually.