
Towards predictive docking at aminergic G-protein coupled receptors.
Procedure described in this paper represents a possible way to predict interactions of antagonists with aminergic GPCRs.
G protein-coupled receptors (GPCRs) are hard to crystallize. However, attempts to predict their structure have boomed as a result of advancements in crystallographic techniques. This trend has allowed computer-aided molecular modeling of GPCRs. We analyzed the performance of four molecular modeling programs in pose evaluation of re-docked antagonists / inverse agonists to 11 original crystal structures of aminergic GPCRs using an induced fit-docking procedure. AutoDock and Glide were used for docking. AutoDock binding energy function, GlideXP, Prime MM-GB/SA, and YASARA binding function were used for pose scoring. Root mean square deviation (RMSD) of the best pose ranged from 0.09 to 1.58 Å, and median RMSD of the top 60 poses ranged from 1.47 to 3.83 Å. However, RMSD of the top pose ranged from 0.13 to 7.33 Å and ranking of the best pose ranged from the 1st to 60th out of 60 poses. Moreover, analysis of ligand-receptor interactions of top poses revealed substantial differences from interactions found in crystallographic structures. Bad ranking of top poses and discrepancies between top docked poses and crystal structures render current simple docking methods unsuitable for predictive modeling of receptor-ligand interactions. Prime MM-GB/SA optimized for 3NY9 by multiple linear regression did not work well at 3NY8 and 3NYA, structures of the same receptor with different ligands. However, 9 of 11 trajectories of molecular dynamics simulations by Desmond of top poses converged with trajectories of crystal structures. Key interactions were properly detected for all structures. This procedure also worked well for cross-docking of tested β2-adrenergic antagonists. Thus, this procedure represents a possible way to predict interactions of antagonists with aminergic GPCRs.
Comparison of studied receptors
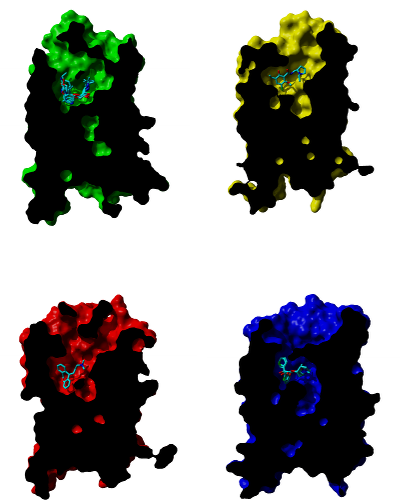
Cross-sections through molecular surface of β‑adrenergic (green), D3 dopamine (yellow), H1 histamine (red) and muscarinic (blue) receptors and their binding sites with bound antagonists. Orientation: extracellular side is up, TM VI and TM VII are in front.