Clock in the choroid plexus in the brain are sensitive to disruption of the daily regime but resistant to inflammatory conditions (15.4. 2024)
In the ventricles of the brain there is a structure called the choroid plexus where cerebrospinal fluid is produced. Recent research has shown that its role is much broader. It also acts as a barrier for the transfer of substances between the blood and the brain, is involved in the active transport of important substances to the brain, in protecting the brain from neuroinflammation and in the removal of metabolic products of the brain, including those that cause neurodegenerative diseases such as Alzheimer's disease and others. Previous studies have shown that the choroid plexus has a robust circadian clock that controls many of its functions to vary throughout the day and night. In our study, we monitored the clock in the choroid plexus in real time under ex vivo conditions by recording rhythmic changes in bioluminescence that correspond to changes in levels of one of the proteins (PER2) essential for the molecular operation of the clock. We found that disrupting the daily regime in the mouse by altering the light-dark cycle (chronodisruption) not only disrupts the clock in the suprachiasmatic nuclei that controls the sleep-wake cycle, but also significantly suppresses the rhythm of the clock in the choroid plexus. A surprising finding was that, unlike other clocks in the body, the clock in the choroid plexus was completely resistant to the pro-inflammatory state induced by the chronodisruption or by the application of lipopolysaccharide. The mechanism of this resistance is likely related to the specific response of the choroid plexus to chronodisruption at the level of glucocorticoid signaling. The results show high sensitivity of the clock in the choroid plexus to disruption of the circadian system. In addition, they showed that operation of the clock is preserved even during proinflammatory state. These results form the basis for a new direction of research into the impact of disrupted function of the clock in choroid plexus on brain homeostasis.
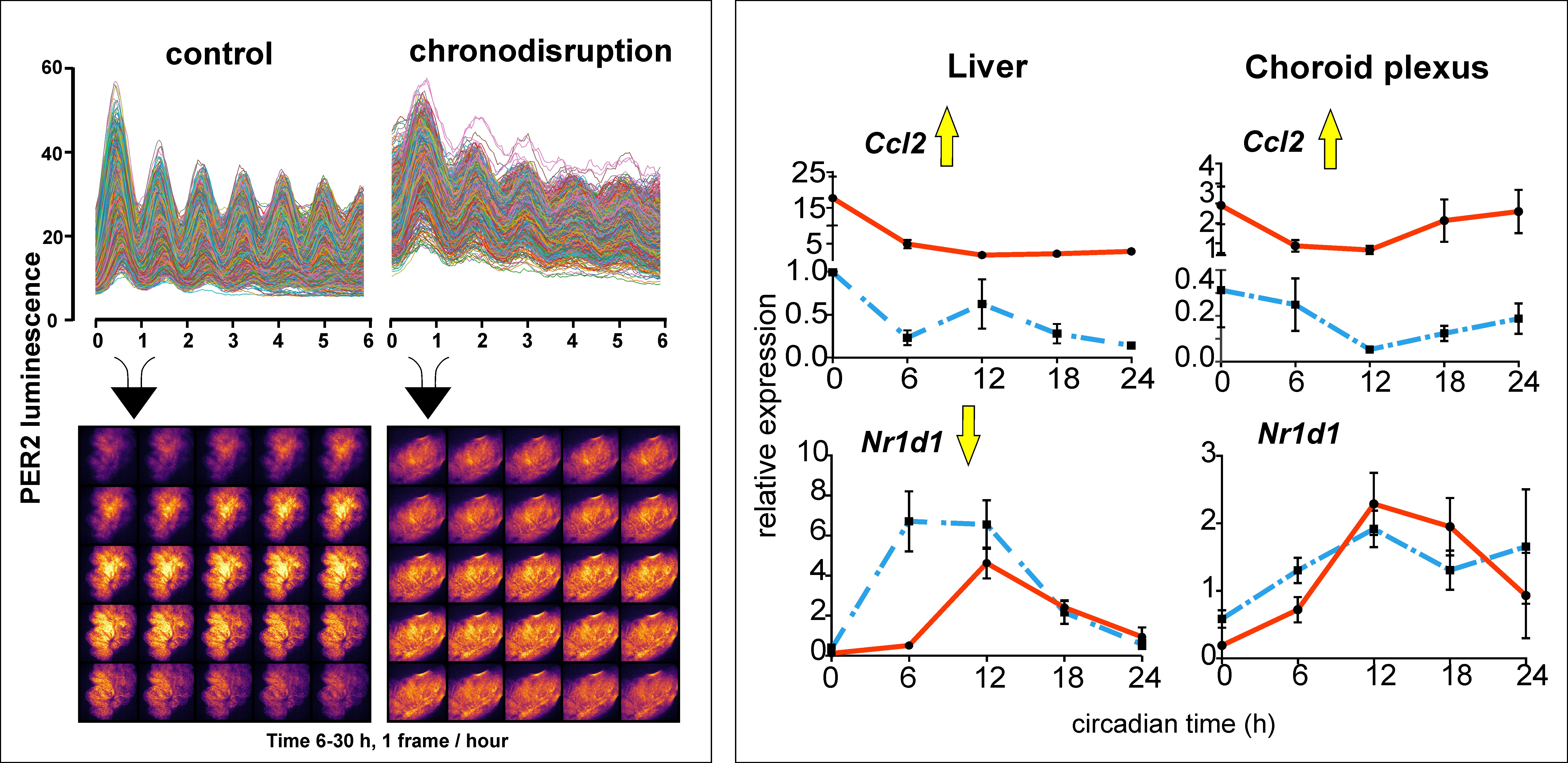
Left: Rhythms of bioluminescence recorded over 6 days with an Olympus LV200 bioluminescence microscope in single cells of the choroid plexus of mPer2Luc mice maintained under control conditions and after exposure to chronodisruption. Right: Differential response of the circadian clock in the liver and choroid plexus to injection of lipopolysaccharide (LPS) in mice. While LPS elicited a pro-inflammatory response (Ccl2) in both tissues, it suppressed the amplitude of rhythmic Nr1d1 clock gene expression only in the liver.
Drapšin M, Dočkal T, Houdek P, Sládek M, Semenovykh K, Sumová A: Circadian clock in choroid plexus is resistant to immune challenge but dampens in response to chronodisruption. Brain Behavior and Immunity. Roč. 117, March (2024), s. 255-269. ISSN 0889-1591. E-ISSN 1090-2139, IF = 15.1 DOI
The ability to transport K+ cations and hence the physiological role of Na+/H+ antiporters is influenced by the composition of their hydrophilic C-termini (31.1. 2024)
It is important for every cell to constantly control the ionic composition of its internal environment (cytoplasm). To cope with the high salt concentrations (e.g. NaCl), cells must be able to eliminate sodium (Na+) cations. On the other hand, the export of potassium cations (K+) from cells is related to the regulation of intracellular pH and membrane potential. Na+/H+ antiporters are among transport systems that ensure the transport of Na+ and K+ out of the cells in exchange for protons in the cells of all eukaryotic organisms, from unicellular yeast to humans. For possible pharmacological intervention in the ion balance, it is necessary to discover, at the molecular level, how the structure of the antiporter impacts its functions.
Na+/H+ antiporters, like most membrane proteins, consist of a part located in the membrane and a part oriented to the cytoplasm - the hydrophilic C-terminus (see figure). Until now, it was assumed that only the transmembrane part determines which ions will be transported by the antiporter. In our new work, we demonstrate that the composition of the hydrophilic C-terminus of the antiporter is also important for determining substrate specificity (especially the ability to transport K+).
For the study, we used a single-cell model organism of eukaryotic cells - the yeast Saccharomyces cerevisiae, in which we expressed the Sod2-22 antiporter from the osmotolerant yeast Zygosaccharomyces rouxii, which efficiently exports only Na+ and Li+ cations, but not K+, from the cells. We found that replacing only one amino acid (introducing a negatively charged residue or removing a positively charged residue) in one of the conserved C-terminal regions (C3) enabled the antiporter to transport K+. Truncation or replacement of the C-terminal part of ZrSod2-22 with the C-terminus from another K+-transporting antiporter (S. cerevisiae Nha1 or Z. rouxii Nha1) also resulted in an antiporter with the capacity to export K+. This work provides a number of new insights into the relationship between the structure and function in the Na+/H+ antiporter family in eukaryotic cells.
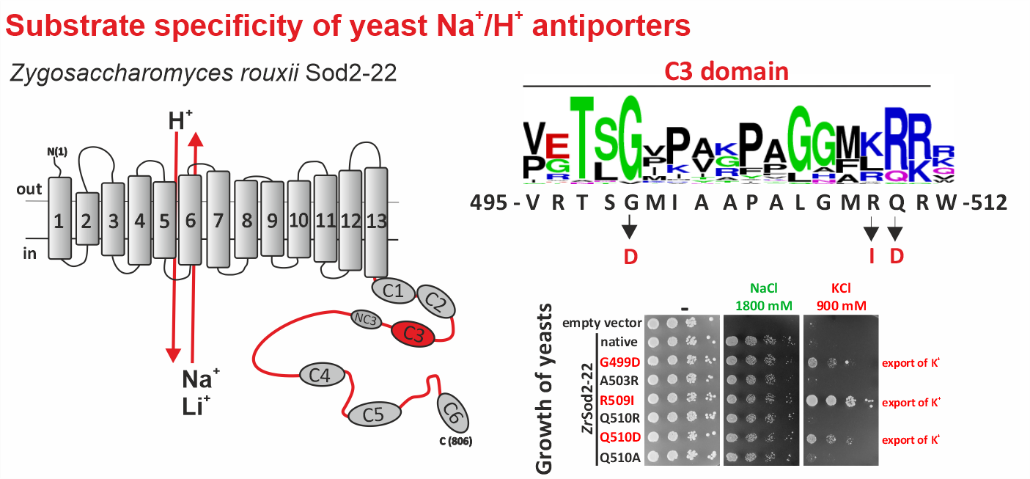
Left: Topological model of Z. rouxii Na+/H+ antiporter Sod2-22 with indicated conserved domains located in the hydrophilic C-terminus. Right: Mutations of particular residues in the conserved domain C3 changed the substrate specificity of the antiporter and enabled it to mediate the export of K+ (demonstrated by the growth of cells expressing these mutated versions in the presence of KCl).
Zimmermannova O., Velazquez D., Papouskova K., Prusa V., Radova V., Falson P., Sychrova H.: The hydrophilic C-terminus of yeast plasma-membrane Na+/H+ antiporters impacts their ability to transport K+. J Mol Biol. 436, 168443 (2024). IF = 5.6 DOI
The "frequency coding" hypothesis provides the classical explanation of the information transmission between nerve cells. This hypothesis relies on the observation that the average rate (frequency) of action potentials increases with stimulation intensity. However, the exact times of action potentials tend to vary even under identical experimental conditions. When evaluating experimental data, or simulated data, we must therefore quantify the variability among multiple trials in order to assess the reliability of frequency coding. The variability of neuronal recordings is usually measured by the Fano factor, whose estimation is considered problematic especially when the amount of data is limited. In this paper, we propose a novel method that allows a precise Fano factor estimation even in situations when the stimulus changes rapidly in time.
Rajdl K, Košťál L: Estimation of the instantaneous spike train variability. Chaos Solitons & Fractals. Roč. 177, December (2023), 114280. ISSN 0960-0779. E-ISSN 1873-2887 IF: 7.8 DOI
How does 14-3-3 protein block CaMKK protein kinase activity? (16.1. 2024)
CaMKK1 and CaMKK2 kinases regulate key physiological and pathological processes such as tumorigenesis, neuronal morphogenesis, synaptic plasticity, transcription factor activation and cellular energy homeostasis, and promote cell survival. We have elucidated the structural basis of the inhibition of both CaMKK kinases by the regulatory proteins 14-3-3. Our results show that binding of the 14-3-3 protein to both CaMKK1 and CaMKK2 prevents their interaction with calmodulin, a signaling molecule that is essential for their activity. Comparison of the structures of the 14-3-3 complexes with CaMKK also revealed that the catalytic center of CaMKK1 is blocked by the C-terminal helices of the 14-3-3 protein. Our findings may help in the development of new drugs targeted to inhibit CaMKK kinases.
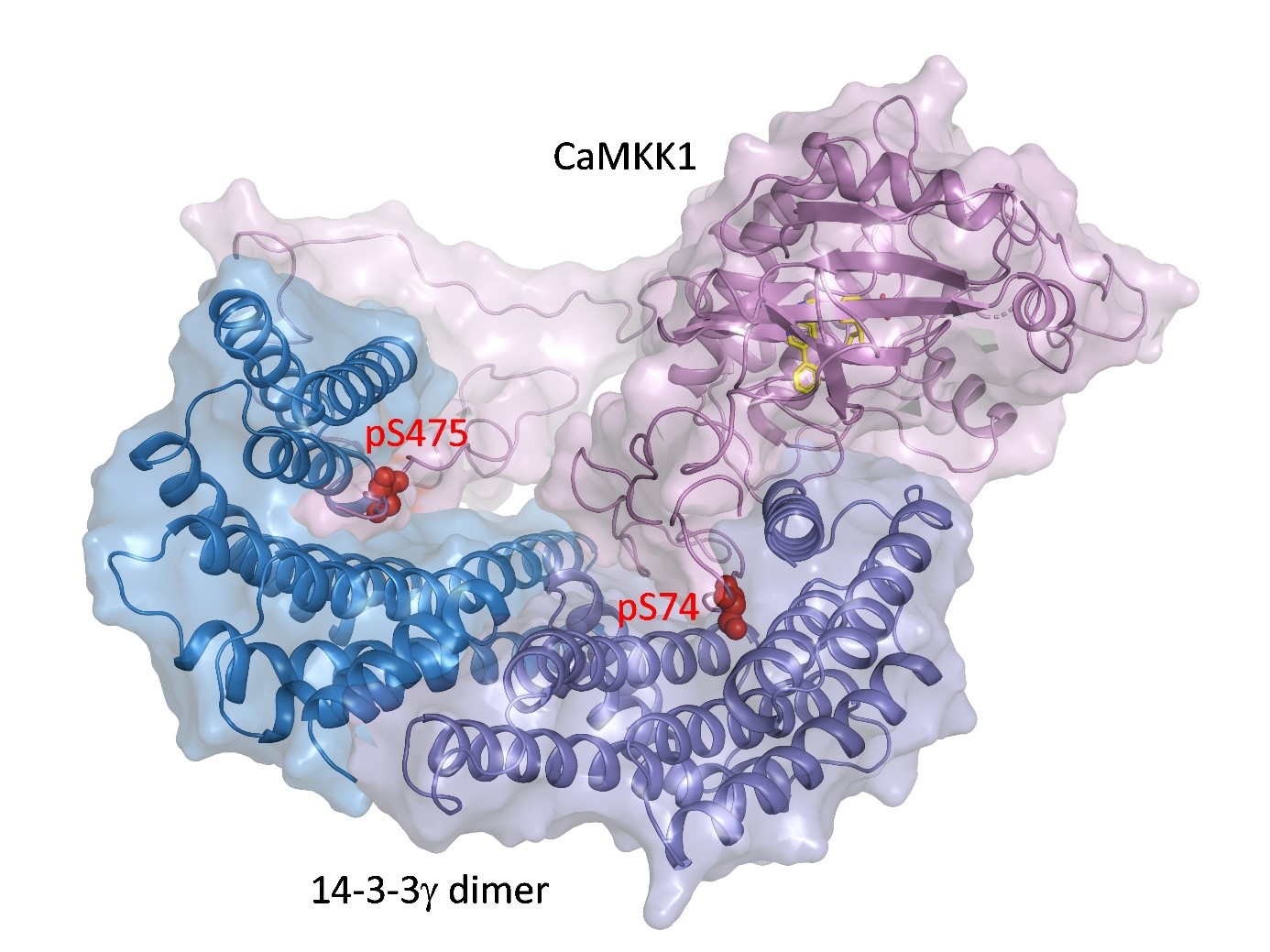
The 14-3-3 protein dimer blocks the catalytic center of CaMKK1 kinase. The kinase active center is shown by the position of the inhibitor (yellow), and the phosphorylation sites of CaMKK1 are shown in red.
Petrvalska O+, Honzejkova K+, Koupilova N, Herman P, Obsilova V*, Obsil T.* 14-3-3 protein inhibits CaMKK1 by blocking the kinase active site with its last two C-terminal helices. Protein Sci. 32 (2023):e4805. ISSN 0961-8368. E-ISSN 1469-896X, IF: 8.000 DOI
+ shared first authorship * shared corresponding autorship
The most common disruption of circadian rhythms in modern society is the so-called social jetlag, i.e. the chronic mismatch between biological time (chronotype) and social time (e.g. waking up according to the alarm clock). However, it is not entirely clear whether social jetlag has any negative effects on health. A large study on a representative adult population of the Czech Republic, conducted by scientists from the Laboratory of Biological Rhythms of the Institute of Physiology of the CAS and published in the journal Sleep, revealed risk factors for the development of cardiovascular and metabolic diseases that are associated with an incorrect sleep schedule.
The authors of the study examined a unique population-representative dataset including 1957 blood samples of adults from across the Czech Republic with different daytime sleep patterns (chronotype). Nine biomarkers (cholesterol, blood lipids, glucose, cortisol, and others) were analyzed in the samples, and an association between social jetlag and biomarkers of cardio-metabolic health (total cholesterol and LDL levels) was found to be statistically significant, especially in people over 50 years of age. The authors also identified new factors influencing social jetlag in the study, such as commuting time to school or work or stress due to lack of time. The study also showed that flexible working hours effectively mitigate social jetlag.
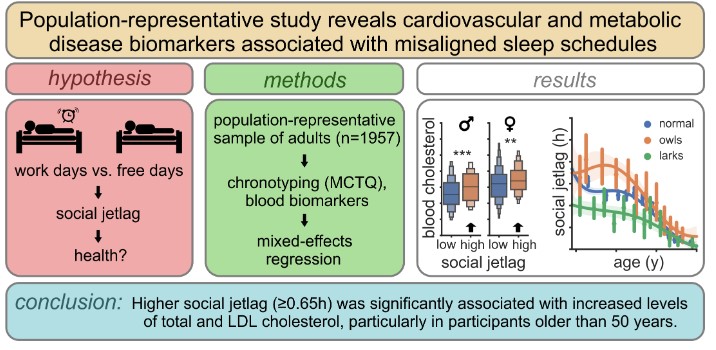
Sládek M, Klusáček J, Hamplová D, Sumová A: Population-representative study reveals cardiovascular and metabolic disease biomarkers associated with misaligned sleep schedules. Sleep, Volume 46, Issue 6, June 2023, zsad037, IF: 5.6 DOI
Load next
