
Vývoj nových postupů pro diagnostiku mitochondriálních chorob
Pro diagnostiku potenciálních pacientů s mitochondriálními poruchami vyvíjíme nové postupy založené na analýze lymfocytů izolovaných z krve. Odběr krve je totiž pro pacienty výrazně méně bolestivý než svalová či kožní biopsie, které se v diagnostice dosud uplatňují.
Dědičná mitochondriální onemocnění a jejich současná diagnostika
Mitochondriální choroby patří k nejzávažnějším dědičným metabolickým onemocněním, která postihují energeticky náročné tkáně, jako jsou srdce, sval, mozek nebo játra, a často se projevují ve velmi raném věku. Tato onemocnění jsou většinou způsobena mutacemi v jaderné DNA, v menší míře pak v mitochondriální DNA (cca 25 %). Zatímco mutace v mitochondriálních genech jsou dobře popsané a jejich diagnostika je relativně snadná, jaderné mutace jsou často neznámé a diagnostika na genové úrovni je v řadě případů neúspěšná. Za těchto okolností je nutné se spolehnout na biochemickou analýzu svalové biopsie nebo kultur fibroblastů založených z kožní biopsie. Získání obou typů vzorků je pro pacienty bolestivé a nezřídka je jejich rodiči odmítnuto.
Cíle a metodika projektu
Ve spolupráci s Klinikou dětského a dorostového lékařství Všeobecné fakultní nemocnice v Praze a Pediatrickou klinikou Fakultní Thomayerovy nemocnice v Praze se proto snažíme zjistit, zda lze pro biochemickou diagnostiku využít lymfocyty izolované z několika mililitrů krve. Při testování vzorků uplatňujeme zavedené metody pro
- kvantifikaci důležitých mitochondriálních enzymů
- proteinové elektroforézy a Western blot
- přímé hodnocení funkce mitochondrií
- měření spotřeby kyslíku na oxygrafu
- cytofluorometrická analýza mitochondriálního membránového potenciálu
Úspěšné řešení projektu umožní šetrnější přístup k vyšetření pacientů, u kterých je vážné podezření, že trpí mitochondriální poruchou.
|
Měření mitochondriálního membránového potenciálu na průtokovém cytometru. |
Výsledek měření – u pacientů s defektem ATP syntázy dochází k nižšímu poklesu membránového potenciálu po přidání ADP. |

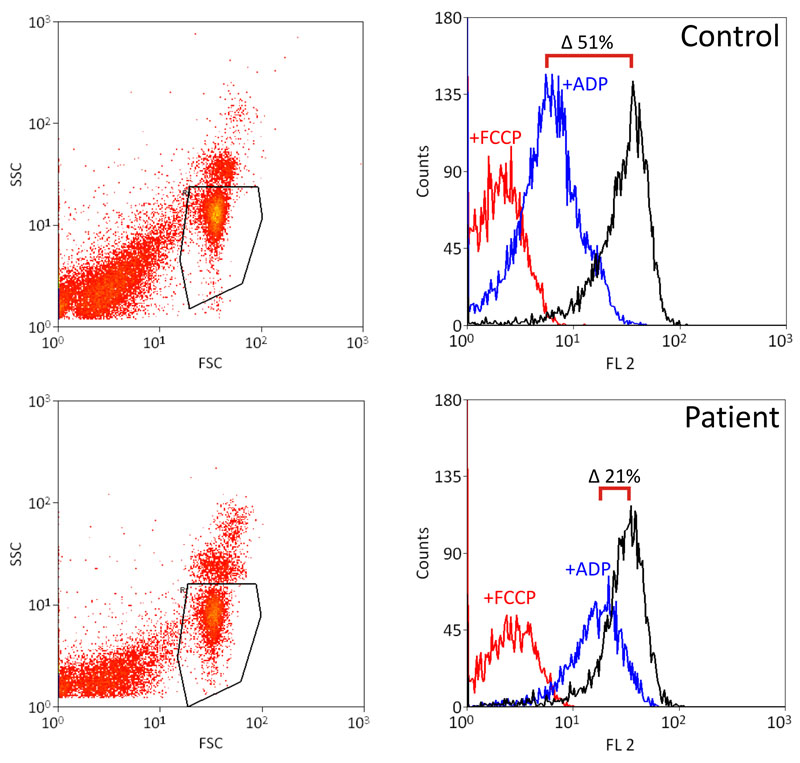
Proteinové komplexy dýchacího řetězce transportují elektrony pocházející z oxidace substrátů na kyslík. Tento proces generuje energii, kterou dané komplexy využijí k pumpování protonů přes membránu z matrix do mezimembránového prostoru. Vzniká tak protonový gradient, který je základem pro mitochondriální membránový potenciál. Ten má jak složku chemickou (rozdíl v koncentraci protonů), tak elektrickou (rozdíl v elektrickém náboji – matrix se jeví jako negativné nabitá). Energie uložená v protonovém gradientu je využita především pro syntézu ATP z ADP a anorganického fosfátu ATP syntázou. ATP syntáza umožňuje transport protonů po gradientovém spádu, což uvolňuje energii pro syntézu ATP. Pokud tedy buňkám přidáme ADP, začne jej ATP syntáza fosforylovat a protonový gradient se snižuje, ovšem ne tolik, jako když přidáme tzv. rozpřahovač (např. FCCP), který protonový gradient na vnitřní mitochondriální membráně úplně zruší.